

在现代生物信息学中,RNA测序(RNASeq)技术已成为研究基因表达的主要工具之一,STAR (Spliced Transcripts Alignment to a Reference) 软件是一款高性能的比对软件,专门用于将RNASeq数据比对到参考基因组上,下面详细介绍基于STAR流程的RNASeq分析步骤:
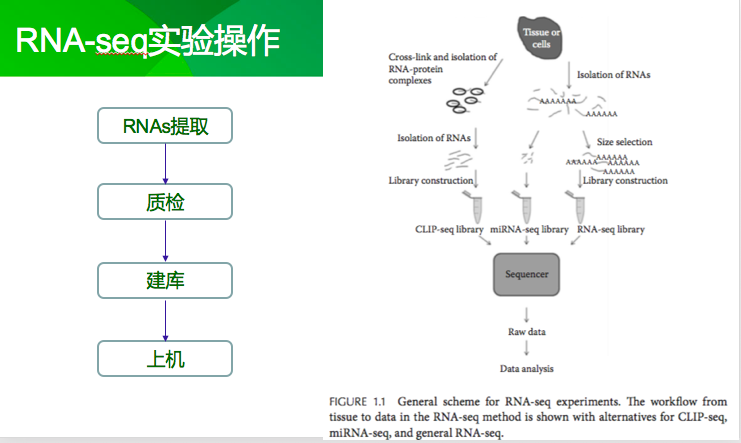
1、准备工作
下载与安装STAR:需要从STAR的官方GitHub页面或其它可靠资源下载最新版的STAR软件,并按照提供的说明进行安装,安装过程通常包括解压文件和设置环境变量。
准备相关软件:除了STAR外,还需要准备其他辅助软件如FastQC、Multiqc、Trimmomatic等,这些软件可以通过conda进行安装,这些工具将用于质量控制和数据预处理。
2、构建基因组索引
生成索引文件:使用STAR时,首先需要为参考基因组生成索引文件,这需要基因组序列文件(FASTA格式)和注释文件(GTF格式),生成索引是进行有效比对的关键步骤。
参数选择:在构建索引时,可以根据需要调整参数,例如设置线程数以加快处理速度,这对处理大规模基因组数据尤为重要。
3、执行Mapping过程
输入数据:将RNAseq数据作为输入,这些数据通常是FASTQ格式的文件,STAR可以接受单端或双端测序数据。
参数设置:在mapping过程中,可以设置多个参数,包括选择之前生成的基因组索引、设置输出文件名和格式、选择是否包含非拼接的读取等,正确的参数设置能确保比对精确和高效。
性能优化:STAR算法通过利用哺乳动物RNAseq数据中的链信息来优化剪切位点的识别,从而提高比对的准确性和速度。
4、质控分析
使用Qualimap:完成mapping后,可以使用Qualimap进行质控分析,这包括检查碱基分布情况,确保数据的均匀性和可靠性。
统计碱基分布:对于reads的每一个位置,统计ATCG四种碱基的分布,通过不同颜色表示不同的碱基比例,帮助快速识别数据中可能存在的问题。
STAR提供了一个高效且准确的解决方案,使得RNASeq数据分析变得快速而可靠,从准备工作到最终的质控分析,每一步都需要细致的注意和优化,以确保得到高质量的分析结果,通过上述步骤,研究人员能够有效地利用STAR软件处理和分析RNASeq数据,从而深入理解生物体的基因表达模式。
原创文章,作者:K-seo,如若转载,请注明出处:https://www.kdun.cn/ask/590282.html
 微信扫一扫
微信扫一扫